Understanding Lysosomal Storage Disorders
Lysosomal storage disorders (LSDs) are a group of rare inherited metabolic diseases caused by defects in specific lysosomal enzymes, transport proteins, or cofactors.
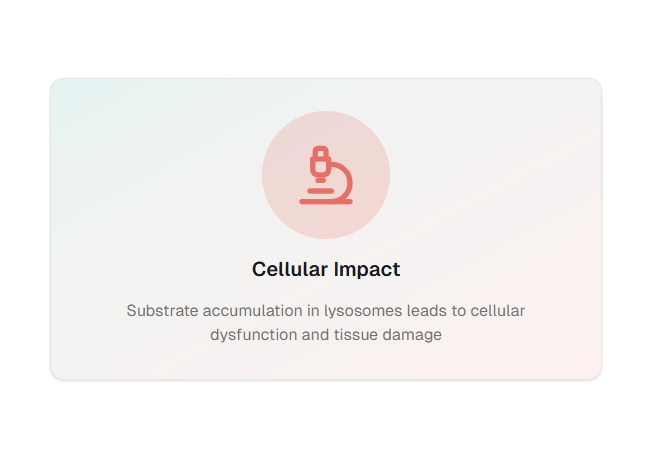
When these enzymes are defective, undegraded substrates accumulate inside cells, leading to progressive multi-organ dysfunction affecting the CNS, musculoskeletal system, cardiovascular system, liver, spleen, eyes, and skin.
More than 50 distinct LSDs have been identified, affecting an estimated 1 in 5,000 to 7,000 live births worldwide.